Smart Nanomaterials Group
General info
Our research interest:
- Development of the Density Functional Theory;
- Spin transport in hybrid structures of single molecule magnets and 2D materials;
- Vertical (van der Waals) and lateral heterostructure of 2D layered materials;
- Ab initio theory of excitations in transition metal dichalcogenides;
- Investigation of the new family of 2D layered materials MXenes, their spintronic, thermoelectric, and optical properties;
- Properties of C-B-N 2d systems;
- Functionalization of 2D materials;
- Interaction of light with matter and plasmonics in 2D systems;
Research methodology and techniques — Multi-scale Atomistic Modeling, involving:
- Ab initio calculations (DFT);
- Molecular Dynamics;
- Monte Carlo Methods;
- Valence Force Field Potentials;
- Tight-Binding Method;
- Non Equilibrium Green Function for transport properties;
- Continuum models (e.g., multi-band k.p method…).
Currently running research projects:
- NCN - OPUS-12, Hybrid structures of single molecule magnets grafted to two-dimensional layered nanomaterials, PI – J. A. Majewski.
- NCN - SONATA-12, Theoretical investigation of structural, electronic, magnetic, and optical properties of van der Waals heterostructure consisting of layered two dimensional materials, PI – Magda Popielska.
- NCN - PRELUDIUM, Stability, ordering, phase diagrams and electronic structures of carbon-boron-nitride 2D hexagonal alloys, PI – Agnieszka Jamróz.
- NCN - SONATA BIS, Research of anti-cancer properties of nano-crystallites two dimensional titanium carbides and nitrides – MXenes Phases, PI – Agnieszka Jastrzębska (Warsaw University of Technology); our group is a–partner in the consortium.
Staff
- prof. dr hab. Jacek A. Majewski
- dr Manel Mabrouk
- dr Magdalena Popielska (Birowska)
- dr Patryk Zaleski-Ejgiert
- mgr Agnieszka Jamróz (Ph.D. student)
- mgr Aleksei Koshevarnikov (Ph.D. student)
- mgr Mariusz Popielski (Ph.D. student)
- mgr Mikołaj Sadek (Ph.D. student)
- mgr Tomasz Tarkowski (Ph.D. student)
- mgr Przemysław Trędak (at present in NVIDIA, California)
- mgr Wojciech Wegner (MISDoMP student, second supervisor: Wojciech Grochala)
- mgr Agnieszka Starobrat (MISDoMP student, second supervisor: Wojciech Grochala)
- Filip Chudzyński (master student)
- Przemysław Zieliński (master student)
- Maciej Marchwiany (associated worker, earlier PhD student, at present in Centre for the Computational Science at ICM UW)
Recent results and publications
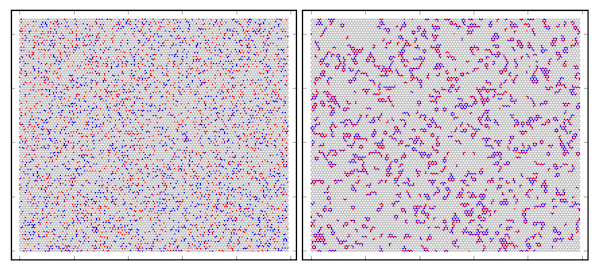
Monte Carlo simulations with Valence Force Field potential for the C0.8B0.1N0.1 alloy system. Initial random positions (left side) and final positions (right side) in the C0.8B0.1N0.1 alloys for simulation at T = 500 K. Red dots denote boron, blue - nitrogen, and grey - carbon atoms. The simulation explains formation of graphene and h-BN domains in the thermodynamic equilibrium conditions.
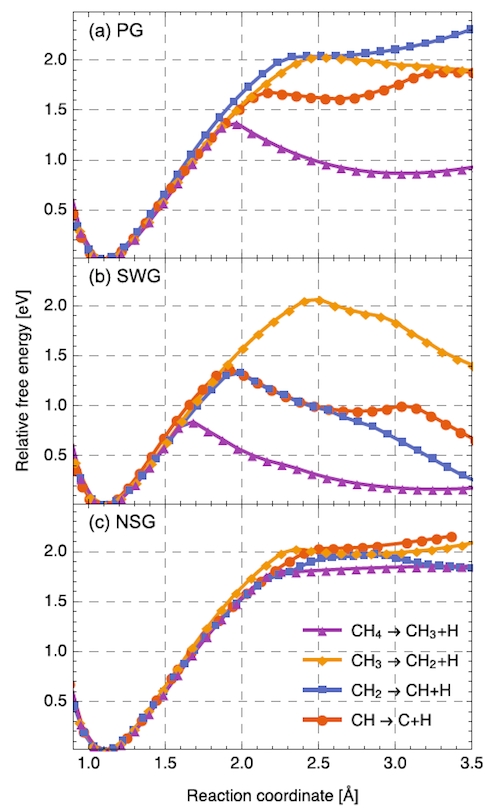
Ab initio molecular dynamics calculations of the dehydrogenation process of methane molecule. Relative free energy profiles of CH4 dissociation steps of (a) pristine graphene, (b) graphene with a SW defect, and (c) graphene with a N substitutional impurity. Curves have been shifted so that zero energy corresponds to global energy minima. The C–H bond distance is the reaction coordinate, for the process CHn —> CHn-1 + H.
Recent publications
- M Wlazło, J. A. Majewski, Free energy landscape of dissociative adsorption of methane on ideal and defected graphene from ab initio simulations, J. Chem. Phys. 148 (2018) 094703
- T. Tarkowski, J. A. Majewski, N. Gonzalez Szwacki, Energy decomposition analysis of neutral and negatively charged borophenes, FlatChem 7 (2018) 42
- Agnieszka Jamróz, Jacek A. Majewski, Ordering effects in 2D hexagonal systems of binary and ternary C-B-N alloys, Computational Materials Science 147 (2018) 113
- M. Birowska, C. Śliwa, J. A. Majewski, Energetic, electronic, and magnetic properties of Mn pairs on reconstructed (001) GaAs surfaces, Phys. Rev. B95 (2017) 115311
- C. Sznajder, N. Huszka, M. Grabowski, J. A. Majewski, Comparative ab initio studies on morphology and stability of the C/BN and SiC/GaN heterostructure interfaces, Mat. Res. Express 4 (2017) 045902
- M. Wlazło, A. Siklitskaya, J. A. Majewski, Ab initio studies of carbon dioxide affinity to carbon compounds and minerals, Energy Procedia 125 (2017) 450
- S. Yastrebov, M. Chekulayev, A. Siklitskaya, J. A. Majewski, R. Smith, Froehlich resonance in carbon nanospiroids and the 2175 A interstellar absorption feature, Nuclear Instruments and Methods in Physics Research B393 (2017) 393
- Magda Birowska, Influence of the different strains’ components on the uniaxial magnetic anisotropy parameters for a (Ga, Mn)As bulk system: A first-principles study, Journal of Magnetism and Magnetic Materials 432 (2017) 390