Grupa Inteligentnych Nanomateriałów
Tematyka badań
Nasze zainteresowania naukowe:
- Rozwój teorii funkcjonału gęstości;
- Transport spinowy w hybrydowych strukturach jednomolekułowych magnesów oraz dwu-wymiarowych materiałów;
- Wertykalne (van der Waalsa) i lateralne heterostruktury dwu-wymiarowych materiałów;
- Teoria ab initio wzbudzeń elektronowych w dichalkogenkach metali przejściowych;
- Badania spintronicznych, termoelektrycznych i optycznych własności nowej rodziny wartwowych dwu-wymiarowych materiałów MXenes;
- Własności dwu-wymiarowych stopów C-B-N;
- Funkcjonalizacja materiałów 2D;
- Oddziaływanie światło-materia oraz plazmonika w systemach dwuwymiarowych.
Stosowana metodologia badań — Wieloskalowe Modelowanie na Skali Atomowej, w tym
- Obliczenia ab initio w ramach teorii funkcjonału gęstości;
- Dynamika Molekularna;
- Metody Monte Carlo;
- Metody potencjału pól siłowych;
- Metoda silnego wiązania;
- Nierównowagowa Funkcja Greena do problem transportu;
- Modele ciągłe (np. wielopasmowa metoda k.p …).
Obecnie realizowane projekty naukowe:
- NCN - OPUS-12, Hybrydowe struktury jednomolekułowych magnesów i warstwowych dwu-wymiarowych materiałów, kierownik – J. A. Majewski
- NCN - OPUS-12, Teoretyczne badania strukturalnych, elektronowych, magnetycznych oraz optycznych własności heterostruktur van der Waalsa złożonych z warstwowych materiałów dwuwymiarowych, kierownik – Magda Popielska
- NCN - PRELUDIUM, Stabilność, uporządkowanie, diagramy fazowe i struktura elektronowa dwuwymiarowych stopów węgiel-bor-azot o strukturze heksagonalnej, kierownik – Agnieszka Jamróz
- NCN - SONATA BIS, Badania właściwości przeciwnowotworowych nano-kryształów 2D karbidków i azotków tytanu - faz MXenes, kierownik – Agnieszka Jastrzębska; Smart Nanomaterials Group jest partnerem w konsorcjum, kierownik ze strony FUW – J. A. Majewski
Pracownicy i doktoranci
- prof. dr hab. Jacek A. Majewski
- dr Manel Mabrouk
- dr Magdalena Popielska (Birowska)
- dr Patryk Zaleski-Ejgiert
- mgr Agnieszka Jamróz (doktorantka)
- mgr Aleksei Koshevarnikov (doktorant)
- mgr Mariusz Popielski (doktorant)
- mgr Mikołaj Sadek (doktorant)
- mgr Tomasz Tarkowski (doktorant)
- mgr Przemysław Trędak (obecnie w NVIDIA, Kalifornia)
- mgr Wojciech Wegner (student MISDoMP, drugi opiekun: Wojciech Grochala)
- mgr Agnieszka Starobrat (studentka MISDoMP, drugi opiekun: Wojciech Grochala)
- Filip Chudzyński (magistrant)
- Przemysław Zieliński (magistrant)
- Maciej Marchwiany (współpracownik, otwarty przewód doktorski, wcześniejszy doktorant, obecnie zatrudniony w Interdyscyplinarnym Centrum Modelowania Komputerowego UW)
Ostatnie rezultaty i publikacje
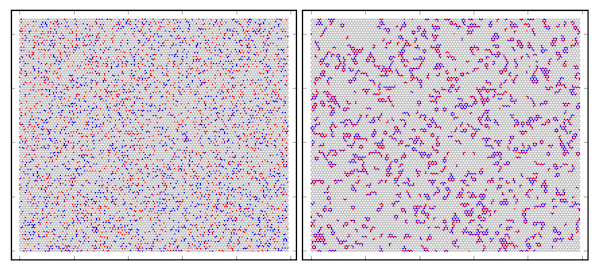
Symulacje Monte Carlo z użyciem metody pól siłowych opisujących oddziaływanie pomiędzy atomami dla dwuwymiarowej warstwy stopu C0.8B0.1N0.1. Przypadkowo rozmieszczone atomy N oraz B w sieci grafenu (lewy panel) na początku symulacji oraz końcowe położenia atomów (prawy panel) po osiągnięciu stanu równowagi termodynamicznej. Czerwone kropki oznaczają atomy boru, niebieskie - azotu, a szare - węgla. Symulacje wyjaśniają tworzenie się obszarów domenowych grafenu i heksagonalnego azotku boru w równowadze termodynamicznej odpowiadającej niskiej temperaturze.
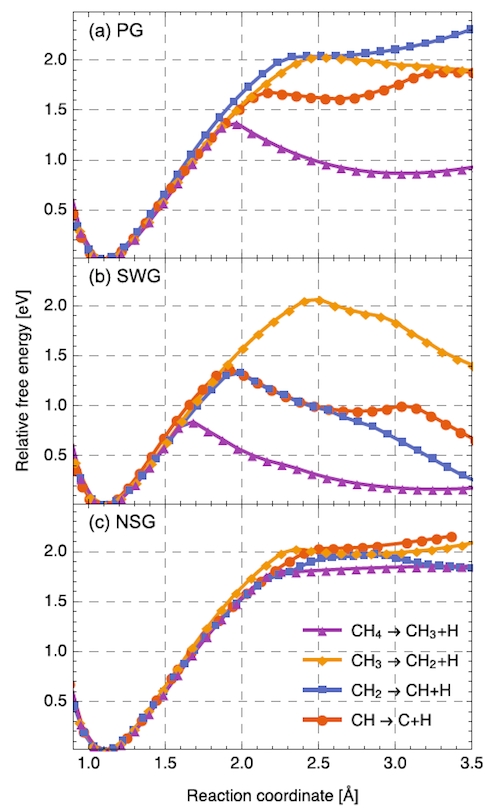
Dynamika molekularna ab initio zastosowana do problemu odwodorniania molekuły metanu przy powierzchni grafenu. Profile względnej energii swobodnej kolejnych kroków odwodorniania molekuły CH4 (a) idealny grafen (bez defektów), (b) grafen z defektem Stona-Walesa, oraz (c) grafen z podstawieniową domieszką azotu w miejscu węgla. Krzywe zostały przesunięte na skali energii, tak, że zero energii odpowiada globalnemu minimum. Parametrem reakcji CHn —> CHn-1 + H jest odległość pomiędzy atomami C–H (długość wiązania).
Ostatnie publikacje
- M Wlazło, J. A. Majewski, Free energy landscape of dissociative adsorption of methane on ideal and defected graphene from ab initio simulations, J. Chem. Phys. 148 (2018) 094703
- T. Tarkowski, J. A. Majewski, N. Gonzalez Szwacki, Energy decomposition analysis of neutral and negatively charged borophenes, FlatChem 7 (2018) 42
- Agnieszka Jamróz, Jacek A. Majewski, Ordering effects in 2D hexagonal systems of binary and ternary C-B-N alloys, Computational Materials Science 147 (2018) 113
- M. Birowska, C. Śliwa, J. A. Majewski, Energetic, electronic, and magnetic properties of Mn pairs on reconstructed (001) GaAs surfaces, Phys. Rev. B95 (2017) 115311
- C. Sznajder, N. Huszka, M. Grabowski, J. A. Majewski, Comparative ab initio studies on morphology and stability of the C/BN and SiC/GaN heterostructure interfaces, Mat. Res. Express 4 (2017) 045902
- M. Wlazło, A. Siklitskaya, J. A. Majewski, Ab initio studies of carbon dioxide affinity to carbon compounds and minerals, Energy Procedia 125 (2017) 450
- S. Yastrebov, M. Chekulayev, A. Siklitskaya, J. A. Majewski, R. Smith, Froehlich resonance in carbon nanospiroids and the 2175 A interstellar absorption feature, Nuclear Instruments and Methods in Physics Research B393 (2017) 393
- Magda Birowska, Influence of the different strains’ components on the uniaxial magnetic anisotropy parameters for a (Ga, Mn)As bulk system: A first-principles study, Journal of Magnetism and Magnetic Materials 432 (2017) 390